The tutorial for webserver of EnrichRBP
Main Functions
Template scripts for automated testing
Please retrieve the template scripts by the link:
Template scripts
Custom Prediction Methods
Input data
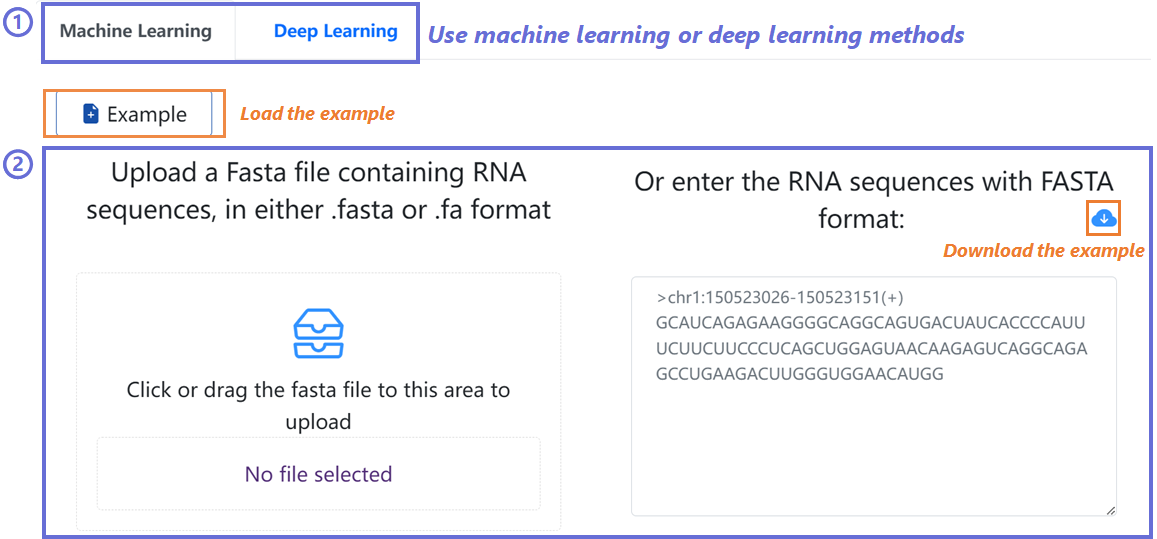
EnrichRBP server is a platform for benchmarking comparison and
interpretation analysis of RNA-binding protein event identification
Methods. The input of the website is RNA sequences, including
circRNA,
linear RNA and RNA in different cells, which can be entered in the
online
input box or uploaded with the standard file format.
Model select
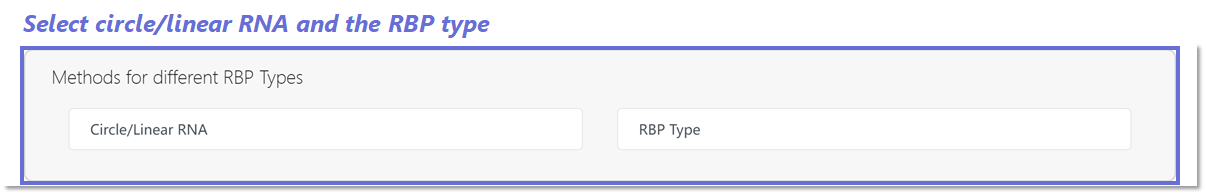
In this step, the users can choose whether the uploaded RNA sequence
belongs to circRNA or Linear RNA, and the RBP type in the database.
Specifically,
37 RBP types interacting with circRNAs were sourced from the study
conducted by iCircRBP-DHN (Yang et al. (2021)).
Additionally, 31 RBP types interacting with linear RNAs were
obtained from HDRNet (Yang et al. (2022)).
Machine Learning Method
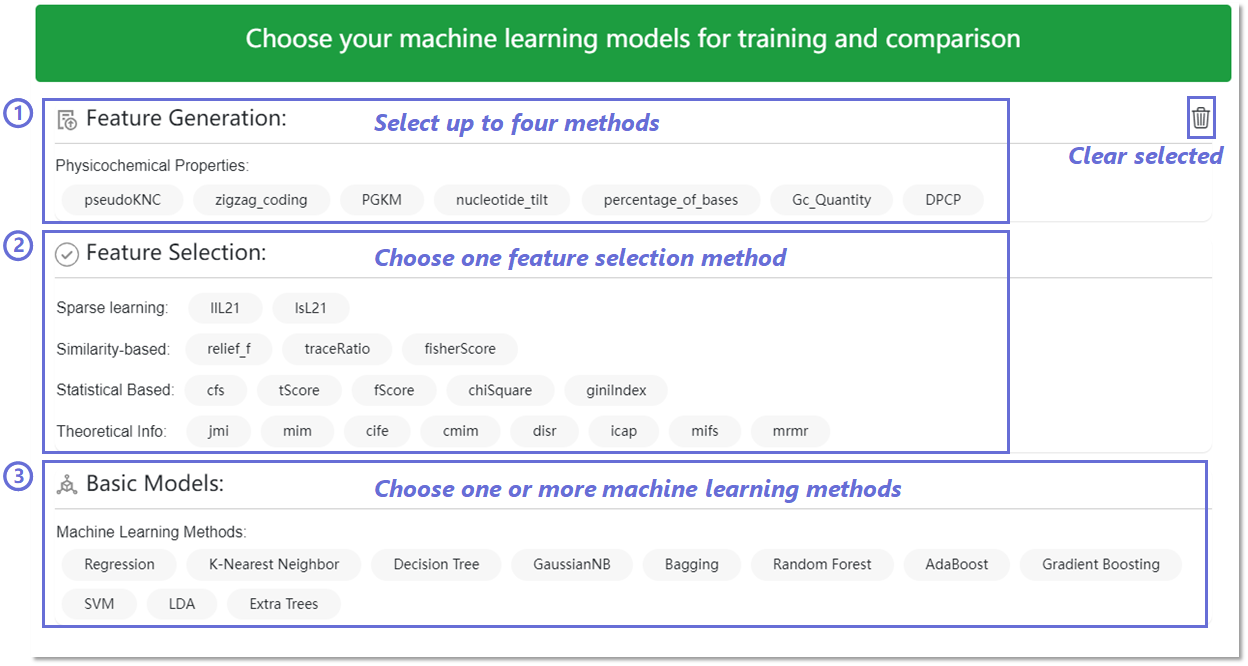
Functional modules including feature generation, feature selection,
sequence prediction, feature analysis and performance visualization
are further added to EnrichRBP to allow users to make initial
classification attempts and understand the subsequent results.
The first module, feature generation, integrates 7 physicochemical
properties. It is worth noting that users can only select up to a
maximum of four. The second module, feature selection, integrates 18
theory-based feature selection methods, including
information-theoretic, similarity-based, sparse learning-based and
statistics-based methods. The third module, sequence prediction,
comprises 11 classical machine learning classifiers (e.g. K-Nearest
Neighbor (KNN), Random Forest (RF), etc). Users can cross-validate
the generated features on the different models. The final module,
feature analysis and performance visualization is designed to
provide users with a visualization of the features and their
performance in various models. If the user has no choice, the
background
will use the default method for training and prediction.
Feature generation methods:
Feature generation methods:
- PseudoKNC (Pseudo K-tuple Nucleotide Composition): PseudoKNC is a method that converts RNA sequences into numerical vectors based on the occurrence frequencies of fixed-length subsequences (k-tuples) of nucleotides. It captures sequence composition information.
- Zigzag Coding: Zigzag coding transforms RNA sequences into numerical vectors by encoding the sequence into a zigzag pattern, which represents the alternation of nucleotide symbols. This method captures certain structural properties of RNA sequences.
- Guanine Cytosine Quantity: This method calculates the proportion or quantity of guanine-cytosine (GC) base pairs in RNA sequences. GC content is often associated with stability and functional properties of RNA molecules.
- Nucleotides Tilt: Nucleotide tilt refers to the angular displacement of base pairs from the helical axis in RNA structures. This feature provides information about the spatial orientation of bases within RNA molecules.
- Percentage of Bases: This method calculates the percentage distribution of individual nucleotide bases (adenine, cytosine, guanine, and uracil) within RNA sequences. It gives insights into the nucleotide composition of RNA.
- Positional Gapped K-m-tuple Pairs: This approach involves extracting gapped k-tuple pairs from RNA sequences, where the tuples are separated by a certain distance. It captures positional information and correlations between nucleotide pairs at specific distances.
- Dinucleotide Physicochemical Properties: This method calculates physicochemical properties (e.g., molecular weight, hydrophobicity, etc.) of dinucleotides present in RNA sequences. It provides insights into the chemical characteristics of RNA molecules.
- Information-theoretic methods:
- JMI (Joint Mutual Information): JMI selects features based on their mutual information with the target variable while considering redundancy among features.
- MIM (Mutual Information Maximization): MIM selects features that maximize the mutual information with the target variable.
- CIFE (Conditional Infomax Feature Extraction): CIFE selects features by maximizing the conditional mutual information with the target variable given the other selected features.
- CMIM (Conditional Mutual Information Maximization): CMIM selects features based on their conditional mutual information with the target variable, taking into account the information provided by already selected features.
- DISR (Double Input Symmetric Relevance): DISR selects features by maximizing the mutual information between feature pairs and the target variable.
- ICAP (Interaction Capping): ICAP selects features by iteratively removing features with the least interaction information with the target variable.
- MIFS (Mutual Information Feature Selection): MIFS selects features based on their mutual information with the target variable while considering redundancy among features.
- MRMR (Minimum Redundancy Maximum Relevance): MRMR selects features that have high relevance with the target variable and low redundancy with each other.
- Similarity-based methods:
- Relief_f: Relief_f evaluates features based on their ability to distinguish between instances of different classes using nearest neighbor-based methods.
- TraceRatio: TraceRatio selects features by maximizing the ratio of between-class scatter matrix to within-class scatter matrix.
- FisherScore: FisherScore selects features by maximizing the ratio of between-class variance to within-class variance.
- Statistics-based methods:
- CFS (Correlation-based Feature Selection): CFS selects features based on their correlation with the target variable while considering the inter-correlation among features.
- TScore: TScore evaluates features based on their t-statistic value, which measures the difference in means of the feature values between different classes.
- FScore: FScore evaluates features based on their F-statistic value, which measures the ratio of between-group variance to within-group variance.
- ChiSquare: ChiSquare selects features based on their chi-square statistic, which measures the independence between features and the target variable.
- GiniIndex: GiniIndex selects features based on their Gini impurity, which measures the degree of class impurity in a dataset.
- Sparse learning-based methods:
- llL21 (L1/L2 Norm Regularization): llL21 selects features by applying L1/L2 norm regularization to induce sparsity in feature weights.
- lsL21 (L2,1 Norm Regularization): lsL21 selects features by applying L2,1 norm regularization to induce group sparsity, encouraging the selection of feature groups rather than individual features.
Deep Learning Method
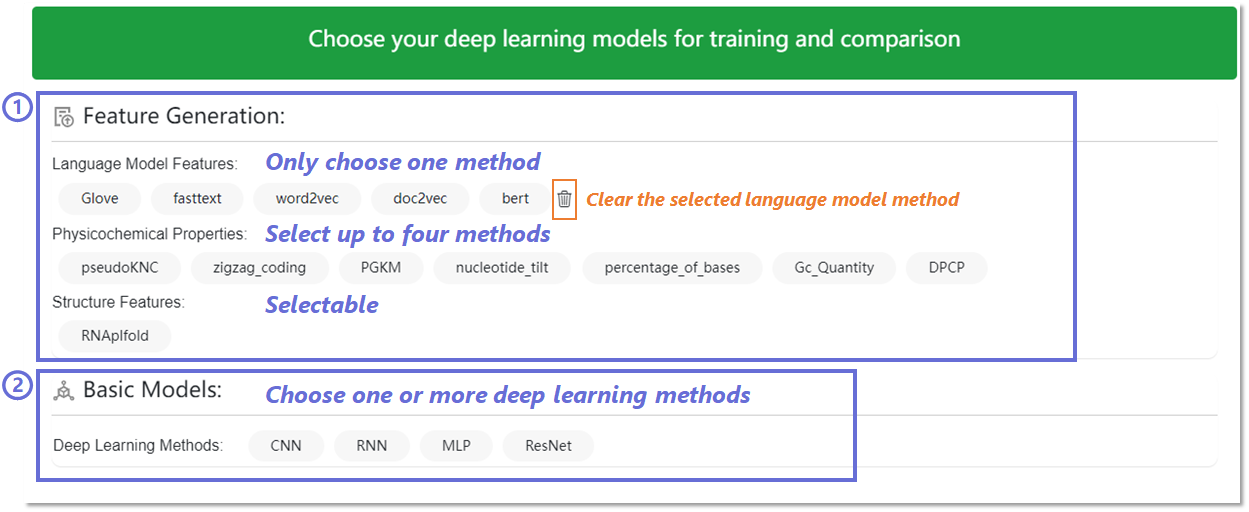
In the deep learning method on the Custom Prediction Methods page,
the first
step is feature generation. We pre-train RNABERT using a
series of masked protein-targeted RNA ’sentences’. In addition, we
generate static local semantics based on RNA sequences using
FastText, GloVe, Word2Vec and Doc2Vec models. Based on these five
language models, we also fragmented the RNA sequences into 3, 4, 5,
and 6 mers, which act as semantic encoders. Users can flexibly
choose these 20 encoding methods. 7 physicochemical properties can
still be selected. We also add RNA secondary structure information
(e.g. RNApfold). In the second step, the above-chosen features are
fed into four deep-learning methods (e.g. Residual Neural Network
(ResNet)). Similarly, if the user has no choice, the background will
use the default method for training and prediction.
- 3, 4, 5, 6mer FastText Semantic Information: Similar to RNABERT, this method uses FastText embeddings to represent RNA subsequences (3, 4, 5, and 6mers) and extract semantic information from RNA sequences. FastText is known for its ability to capture subword information effectively.
- 3, 4, 5, 6mer GloVe Semantic Information: GloVe (Global Vectors for Word Representation) is another word embedding technique used for generating semantic information from RNA subsequences (3, 4, 5, and 6mers). It captures the global co-occurrence statistics of words in a corpus to create embeddings.
- 3, 4, 5, 6mer Word2Vec Semantic Information: Word2Vec is a widely used word embedding technique that learns continuous vector representations of words based on their context in a large corpus of text. Here, it's applied to RNA subsequences (3, 4, 5, and 6mers) to extract semantic information.
- 3, 4, 5, 6mer Doc2Vec Semantic Information: Doc2Vec extends Word2Vec to learn vector representations of entire documents. In this case, it's applied to RNA sequences to generate semantic information based on embeddings of entire sequences and their subsequences (3, 4, 5, and 6mers).
- Secondary Structure Information: This method involves predicting the secondary structure of RNA sequences, which includes features such as stem-loop structures, hairpins, and base pairings using RNAplfold. These features provide insights into the structural properties of RNA molecules.
Submit
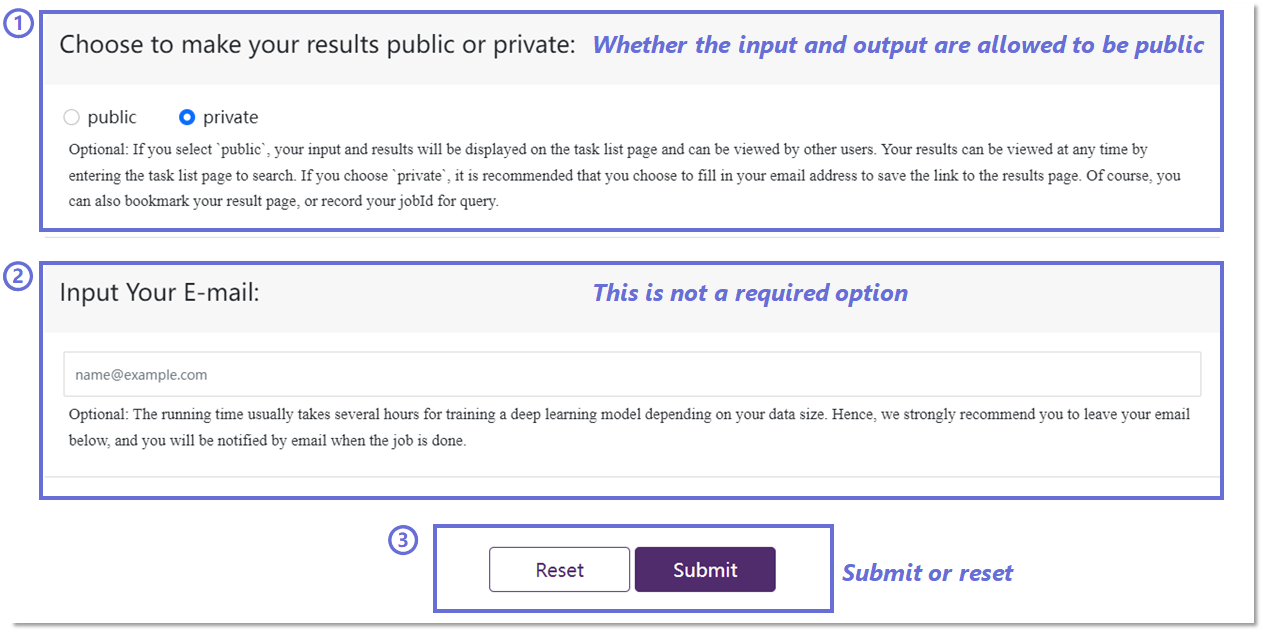
To enhance the accessibility of historical predictions for users, we
allow you to choose whether to make the results public or private.
Opting for the public setting will display the results on the task
list page while selecting private will keep the results hidden.
To ensure timely access to model prediction results, we recommend
you provide your email addresses for the delivery of the result
link, although this step is optional.
Algorithms and RBP type selection for CircRNA-RBP pages
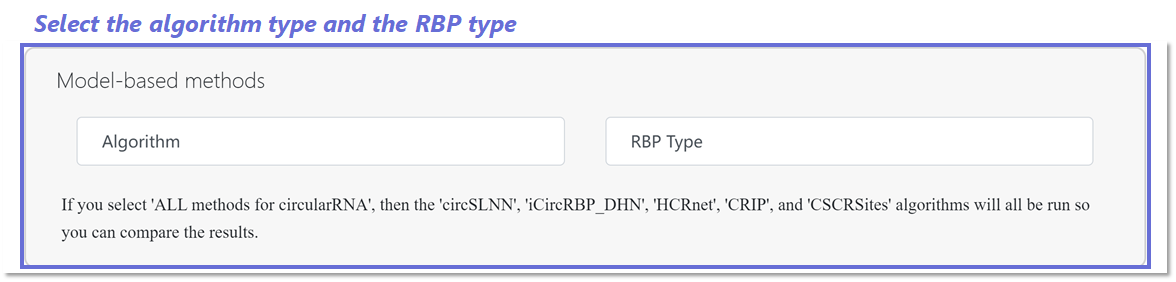
In CircRNA-RBP pages, you can choose from 5 algorithms (e.g. circSLNN,
iCircRBP-DHN, HCRNet, CRIP, and CSCRSites) as prediction methods. If
you select 'ALL methods for circularRNA', then the above algorithms will
all be run. You then need to select the input RBP type.
- circSLNN (https://github.com/JuYuqi/circSLNN) is driven by pre-trained RNA embedding vectors and a composite labelling model for identifying specific locations of RBP-binding sites on the circular RNA.
- CRIP (https://github.com/kavin525zhang/CRIP) consists of a stacked codon-based encoding scheme and a hybrid deep learning architecture, in which a convolutional neural network (CNN) learns high-level abstract features and a recurrent neural network (RNN) learns long dependency in the sequences.
- CSCRSites is a deep learning-based approach to identify cancer-specific circRNA-RBP binding sites using only nucleotide sequences as input.
- iCircRBP-DHN (https://github.com/houzl3416/iCircRBP-DHN) proposes a new coding scheme by integrating CircRNA2Vec and K-tuple nucleotide frequency patterns to represent different degrees of nucleotide dependencies. It also utilises deep multi-scale residual networks, as well as bi-directional gated recursive units (BiGRUs) with self-attention mechanism, to extract both local and global contextual information.
- HCRNet (https://github.com/yangyn533/HCRNet) is an end-to-end framework for identifying circRNA-RBP binding events. To capture hierarchical relationships, it incorporates multiple sources of biological information to represent circRNAs, including various natural language sequence features. In addition, a deep temporal convolutional network incorporating a global expectation pool is included to exploit potential nucleotide dependencies in an exhaustive manner.
Algorithms and RBP type selection for Linear RNA-RBP page
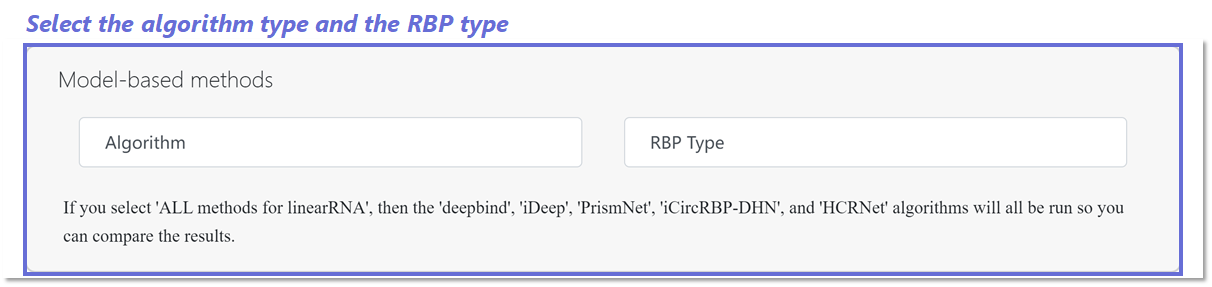
On the Linear RNA-RBP page, you have the option to choose from five algorithms (such as DeepBind, iDeep, PrismNet, iCircRBP-DHN, and HCRNet) for prediction methods. If you select 'ALL methods for linear RNA', all the mentioned algorithms will be executed. Following this, you will need to specify the input RBP type.
- DeepBind (https://github.com/jisraeli/DeepBind) is a CNN based deep learning model that predicts RBP binding sites based on only RNA sequences.
- iDeep (https://github.com/xypan1232/iDeep) is proposed and leverages a novel hybrid CNN network and deep belief network to predict the RBP interaction sites and motifs on RNAs by converting the original data into a high-level abstraction feature space using multiple layers of learning blocks, where the shared representations across different domains are integrated.
- PrismNet (https://github.com/kuixu/PrismNet) is a recent study that developed a convolutional neural network (CNN) based deep learning approach, which effectively incorporates in vivo RNA structure data and RNA-binding protein (RBP) binding data to make precise predictions of RBP binding sites. This method applied an “attention” strategy to precisely identify RBP binding nucleotides. Notably, PrismNet is the first tool designed for dynamic prediction tasks.
Algorithms and RBP type selection for RNA-RBP in Cells page

In RNA-RBP in Cells page, users can choose which algorithm to use (e.g. HDRNet
and PrismNet), then select the cell type (e.g. K562) and choose the
RBP type (e.g. AARS) to combine with the linear RNA.
Regarding data sources, a dataset encompassing 261 cell line RBP
binding sites was acquired from Zhu et al. (2023),
spanning across multiple databases. Notably, this dataset includes
binding sites of 172 RBPs identified
using the same tagging technique in cell lines such as K562, HepG2,
HEK293, HEK293T, HeLa, and H9.
- HDRNet is an end-to-end deep learning-based framework that accurately predicts dynamic RBP binding events under various cellular conditions. HDRNet outperforms other state-of-the-art models on 261 linear RNA datasets from eCLIP and CLIP-seq.
Task List
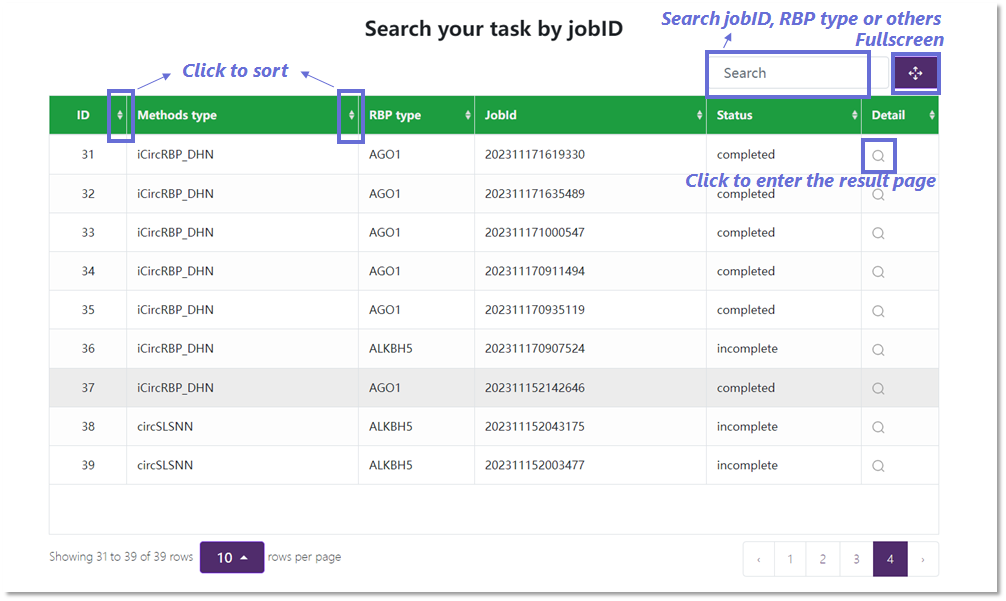
In the task list interface, you can view the public task completion status and task results. To facilitate user
operations, we have also added sorting and search functions.
Result report
Case Study One
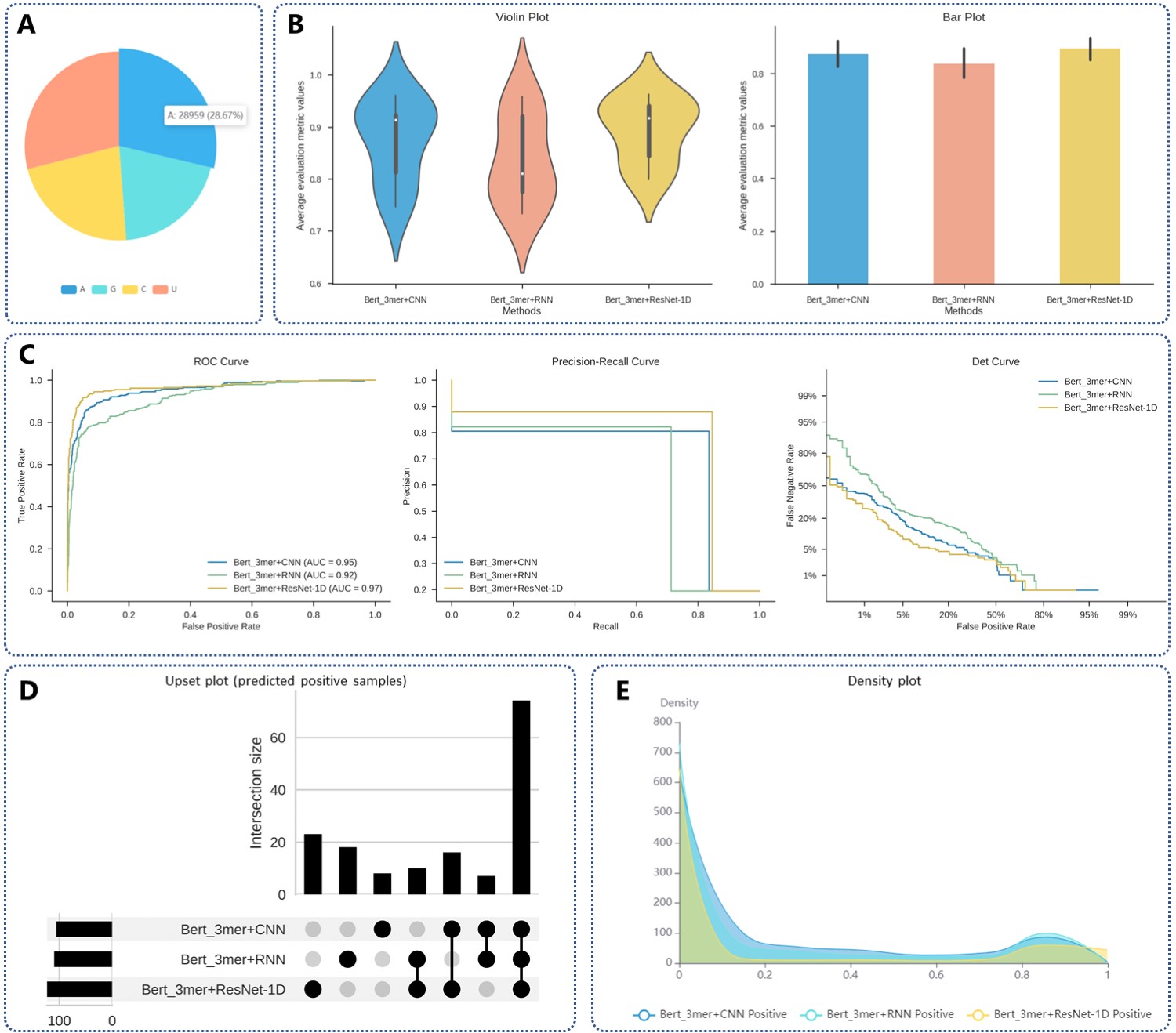
Analysis of results using customized methods to predict linear RNA combined with QKI.
(A) Pie chart of the number and proportion of A, G, C, and U in the user input sequence.
(B) Violin and bar plots depicting the performance of various methods. In the violin plot, the value represents eight evaluation indicators: AUC, ACC, Recall, F1-Score, MCC, Precision, Specificity, and AP. The bar chart displays the average evaluation indicator value, calculated as the mean of these eight indicators.
(C) ROC, PR, and Det curves for each model.
(D) The upset plot to express the relationship between the predictions of different models (only samples predicted as binding sites are shown).
(E) The density plot illustrates the distribution of results from various deep-learning models used to predict whether an RNA sequence is an RBP binding site.
Case Study Two

Analysis of results using Custom Methods to predict linear RNA binding to hnRNPC-2.
(A) Performance comparison among different models in terms of AUC, ACC, Recall, F1-Score, MCC, Precision, Specificity (SP), and AP values.
(B) The ROC curve of different machine-learning models.
(C) Learning curves for individual models, including training and cross-check scores.
(D) The discrimination threshold visualization of precision, recall, f1 score, and queue rate concerning the discrimination threshold of one classifier.
(E) SHAP feature importance analysis using a beeswarm plot.
(F) The heatmap plot for illustrating the cumulative impact of features on predictions from the perspective of multiple features.
(G) Waterfall plot for explanations for individual predictions. (H) Beeswarm plot for the interaction between feature 3 and the other feature.
Saliency maps
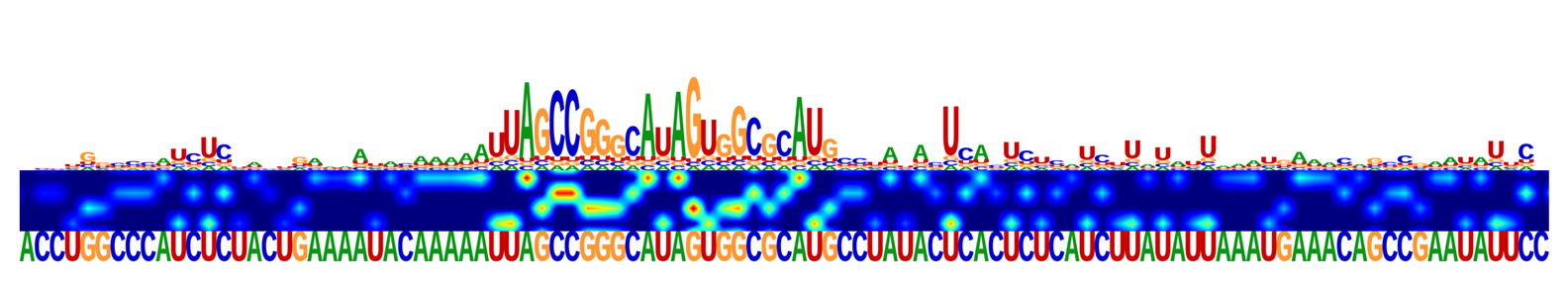
For the prediction of RNA-RBP interactions within different cell lines, we generate saliency maps that identify motifs and interpret crucial features across the entire sequence.
The saliency maps of the dynamic prediction of RBP, where the top strip plots the potential binding motifs;
the second strip is the heatmap of the sequence attention scores and the third strip indicates the specific sequences.
Motif analysis
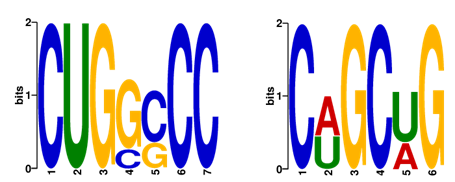
This figure shows the results of a motif discovery analysis using MEME. The analysis was designed to identify conserved sequence motifs
within a set of biological sequences. Each motif represents a recurring pattern that may be an RBP binding site. Discovered motifs are
represented graphically and each motif is depicted in the form of a sequence logo. Specifically, We filtered RNA sequences predicted as
RBP binding sites, retaining the top two ranked motifs predicted by the meme method with p-values < 0.05 and lengths of 6-7 nucleotides.
It is worth noting that the higher the number of predicted real sequences, the higher the confidence of the extracted motif.
For an explanation of the other output graphs, please see the supplementary material